thank you for your quick reply. I was able to execute the batch processing of BiophysicalOP operator in command line GPT tool, the -q option really did the job and now the retrievals are running of all cores.
However, I’m still struggling with the Resample operator in GUI mode - it seems, that the GUI batch processing uses a new bullet switch (SNAP 5.0) instead of cells with values (SNAP 4.0) for setting the resampling parameters. For some reason, my preference in resampling all bands to 20 m resolution (i.e. Target resolution 20) is not working, since the GUI puts also the Target width and Target height 100 there. This produces an error message: if targetResolution is set, targetWidth, targetHeight, and referenceBandName must not be set . I was able to overcome this only by manually editing the XML file (i.e. deleting the targetWidth and targetHeight tags - see the previous XML graph attached) and executing the Graph via command line. Perhaps this is a bug in SNAP 5.0 related to new bullet switch option for resampling? Or am I doing something wrong?
I couldn’t reproduce this behaviour. The 100 pixels in width and height are replaced as soon as I have selected a product. Maybe this is the reason why you observer the problem. Maybe you need a product.
I have also a question about the biophysical operator. I have already tested the option with the .xml file (gpt operator.xml path_to_xml -q 160 -PInput=“path_to_xml” -POutput=“output.tif”), as well as the gpt command (gpt BiophysicalOp -PcomputeCab=true -PcomputeCw=true -PcomputeFapar=true -PcomputeFcover=true -PcomputeLAI=true -q 160 -Ssource=“path_to_resampled” -f GeoTIFF-BigTIFF -t output.tif) on a resampled product (extracted via another xml). Is there a performance difference between the two commands? Furthermore, I did not notice a time difference for the computation of 3 vs 5 indices. My main problem is that it takes too long even in 20+ cores machine. Which is the preferred method (command) in order to run the biophysical operators, and also are there any expected average times as benchmark (beacause I want to run for multiple products)?
You see probably no performance difference because the algorithms are not very complex and fast. That your processing is slow has probably other reasons.
Some thoughts for improving the processing:
You should not use the -q options, at least not setting it to such a high value. By default, the number of parallel threads is equal to the number of cores available. Which is a good choice usually.
When having 160 threads also the data is allocated for 160 tiles and tis might overload your memory.
Regarding memory. Probably you have also a lot available in your machine.
Change in the gpt.vmoptions the heap space settings to
-Xmx16G
or more. I would suggest 70%-80% of the available RAM.
You can also set the cache size used by gpt.
Use the -c option and set it to 70%-80% of the heap space.
Hi,
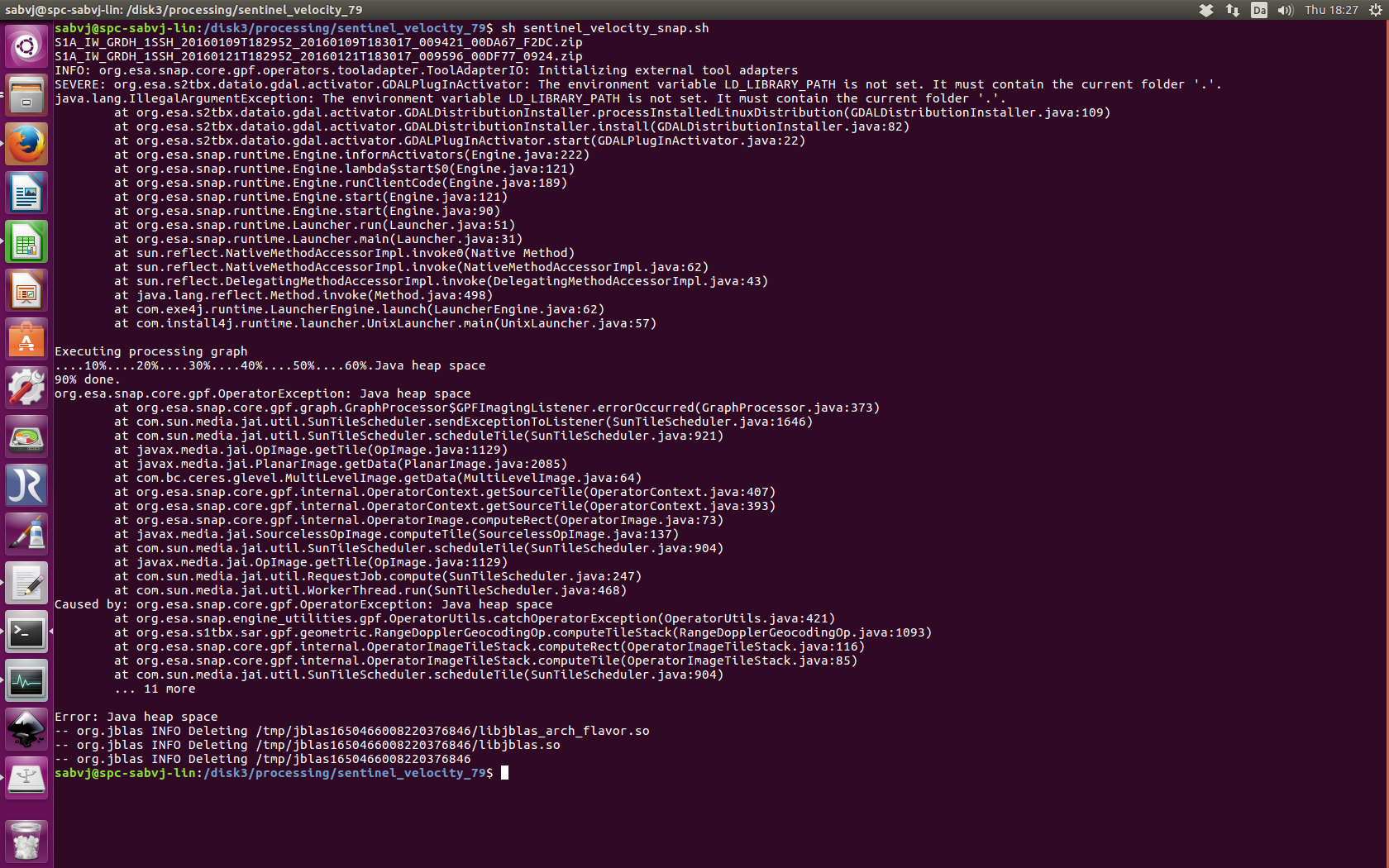
Even though I used your recommended settings using -q 120, I still get heap space error.
I use it like that gpt “$wd/test20170615_glacier_velocity_commandline.xml” -q 120 -Pmaster="$wd/$master" -Pslave="$wd/$slave" -Poutput="$wd/$output.tif"
I also wonder why it didn’t happen last time when I processed the scene couple of days ago.
Please suggest.
I changed the memory settings. I opened /snap/bin/gpt.vmoptions and uncomment the memory setting text. Now its Xmx16G.
Afterwards I run gpt “$wd/test20170615_glacier_velocity_commandline.xml” -q 120 -Pmaster="$wd/$master" -Pslave="$wd/$slave" -Poutput="$wd/$output.tif"
Please see the snapshot what I got.
Hi,
Now I have increased the heap space by Xmx28G (gpt.vmoptions).
I have also increased the cache size by changing snap.properties
snap.jai.tileCacheSize = 24576
snap.jai.defaultTileSize = 512
I wonder snap.parallelism=1 is commented by # default. Is that alright ?
Anyway, after doing these settings, the graph processing is still going on (more than 2.30 hours). Couple of days ago, when the graph processing was normal, it took 1.15 hours. I wonder what really changed. Its the same processing graph, same dataset.
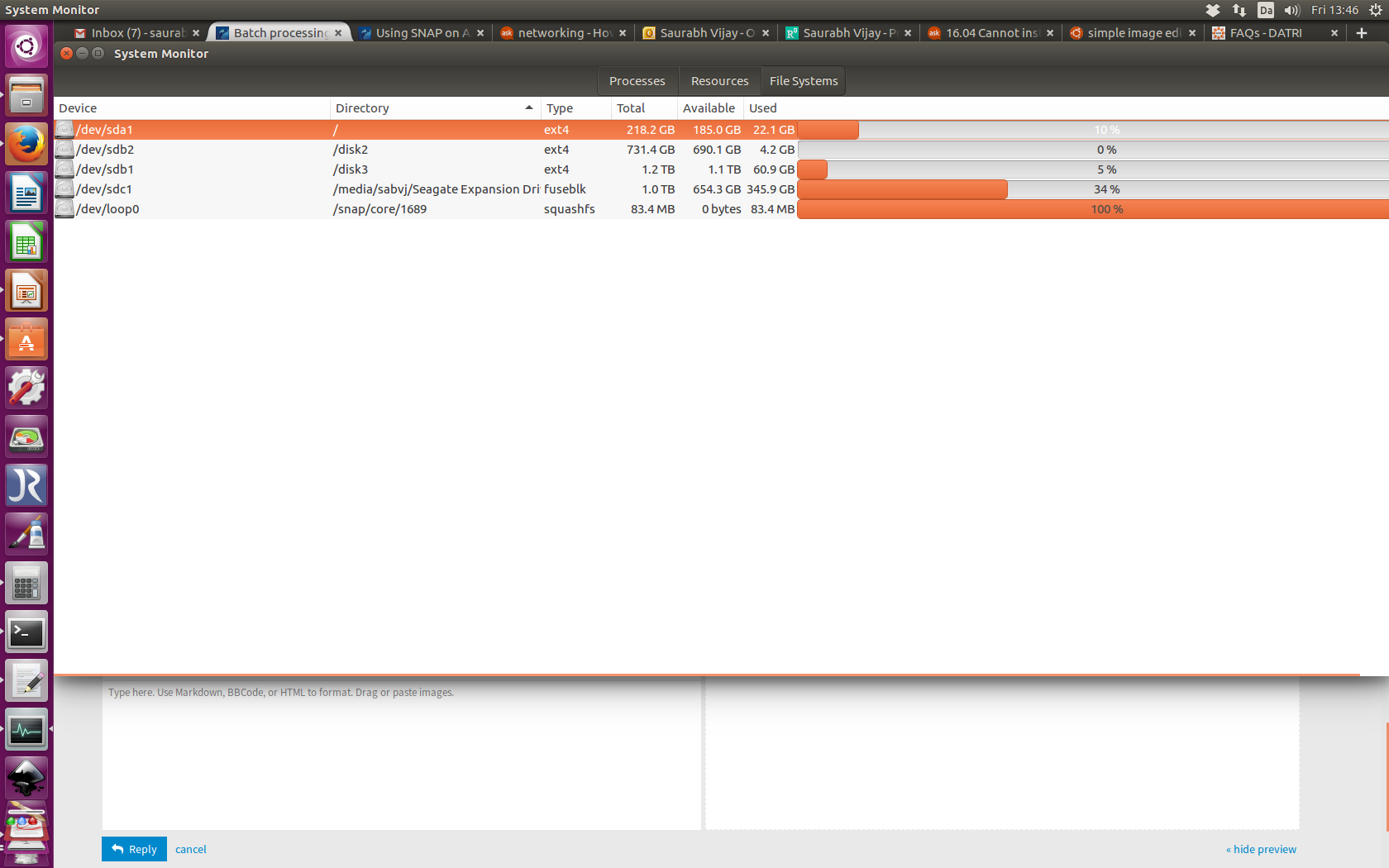
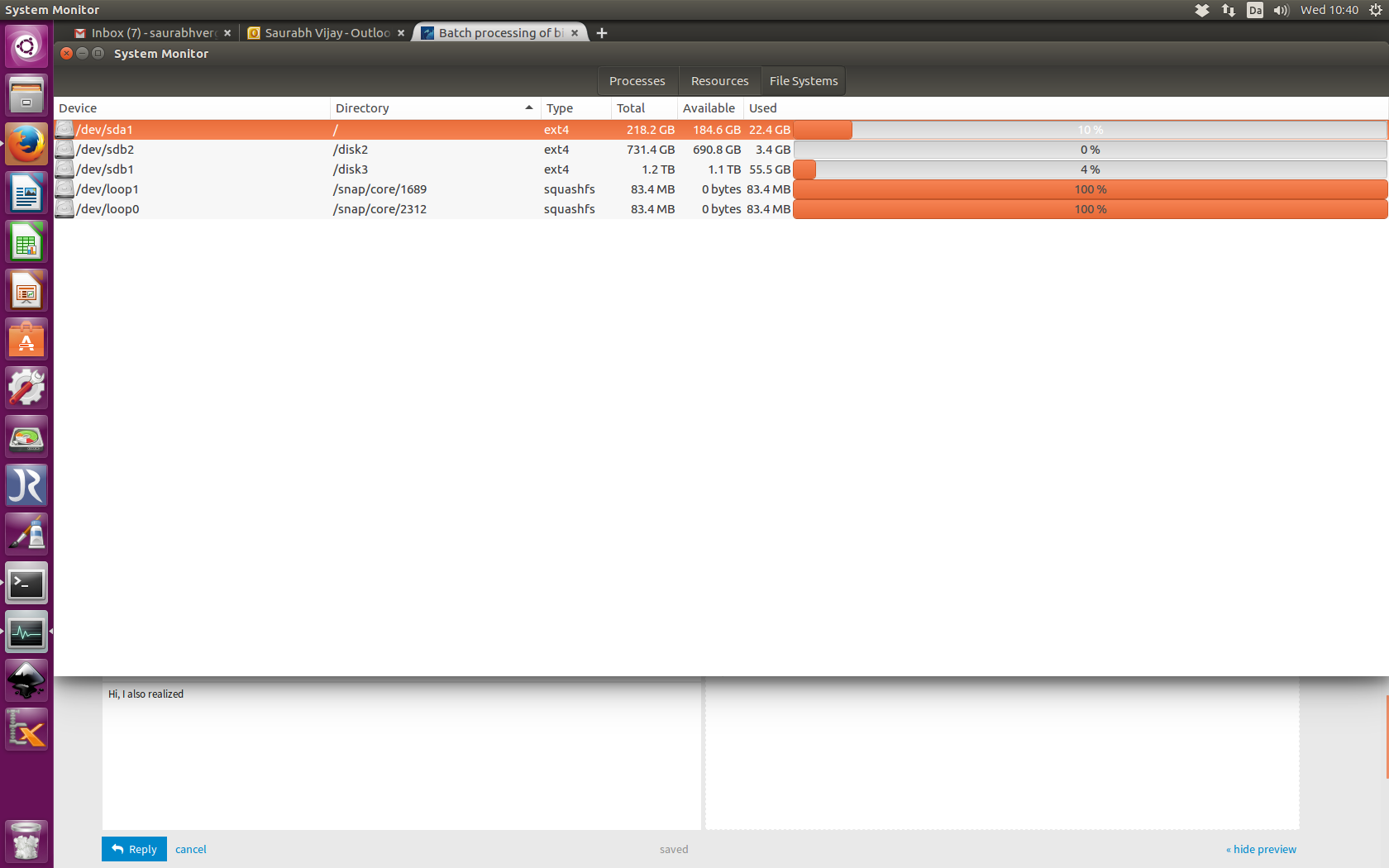
Does it have anything to do with snap/core/1689 ? See attachment.
Hi there,
I tried the settings, please see my above post. It is still not working. I even processed the graph using user interface, it also shows “Java heap scape” error. Please suggest. May be its a bug.
Yes, this is correct. If the value is not set, snap uses the number of cores available to set the parallelism.
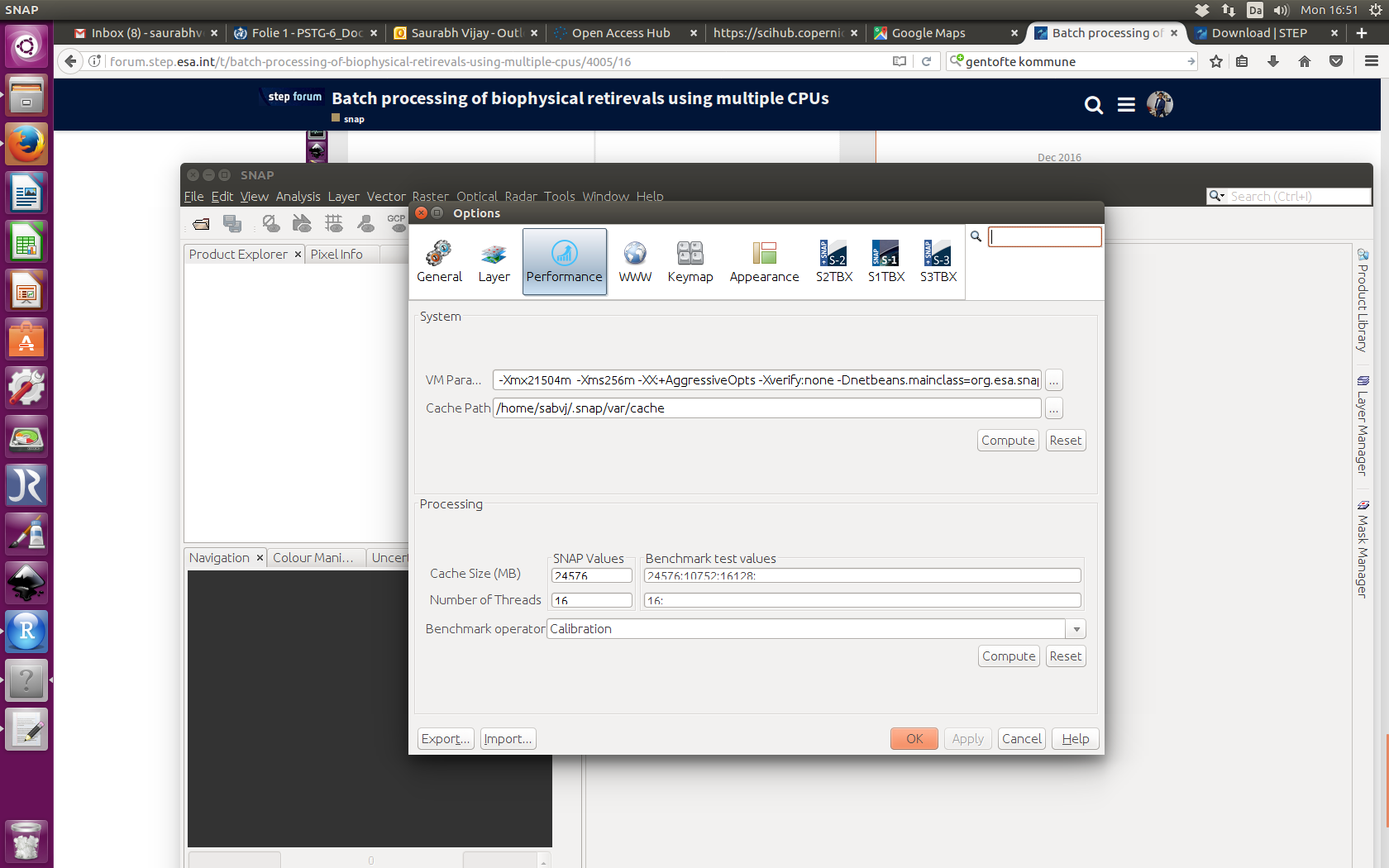
Have you tried your graph with the Performance Tool (Tools -> Options -> Performance)? Have a look what it will suggest. The run might take a while.
The “Java heap scape” error indicates that 28G is still not enough.
Are you doing the Terrain-Correction for both products in one graph? If so, it might be better to split this into two graphs.
You can try to call gpt --diag This will show you the effective configuration.
Hi @marpet, somehow it is still not working. It’s a mystery for me now.
I have a strong feeling that it has something to do with allocated memory. Have a look here. snap/core/ size is full
Dear @marpet,
I confirm the problem when using the Resample and the BiophysicalOp in a single graph. Processing them in a single graph utilises one core, even if -q and -c options are correctly set (from gpt --diag).
When biophysical processor is called from SNAP or as single operator in gpt (gpt BiophysicalOp) utilises multiple cores, when BiophysicalOp is called in gpt from a graph (containing only Read + BiophysicalOp + Write operators) utilises only one core.
Hope this can be solved.
Sorry for an unspecific request but on november the 28th ,I upgraded Snap6 0 ,and everything went wrong
the update failed
Now the software crashed : i can’t do anything
Am I the only one
HELP
thanks