by cleaning you mean deleting I suppose?
I am working on Linux.
I have the following folders in my /home/user/.snap
auxdata epsg-database etc graphs product-library system var
shall I delete them?
I do not know what would be the AppData folder for Linux. Could you advise?
I managed to reproduce on both Windows and Linux, before I have forgotten to change the subset extent when changing the product
What I have also noticed: if you change the formatName to GeoTIFF (and the extension of the output to tif instead of dim), it works.
For example, in your graph, change the Write node to:
You haven’t changed the output extension, it is still .dim (S2A_MSIL2A_20220206T154431_N0400_R011_T19TCJ_20220206T202105_resampled_subset.dim). Try change it to S2A_MSIL2A_20220206T154431_N0400_R011_T19TCJ_20220206T202105_resampled_subset.tif
correct for the file name, thanks.
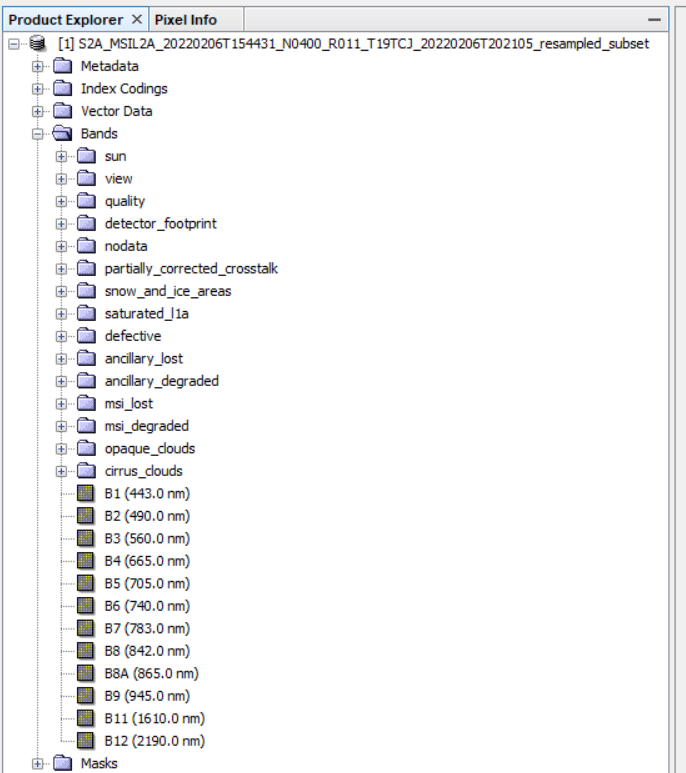
I used to have 2 outouts from this graph:
one dim, now a tif file: S2A_MSIL2A_20220206T154431_N0400_R011_T19TCJ_20220206T202105_resampled_subset.tif
one datafile containing all the S2 bands:
S2A_MSIL2A_20220206T154431_N0400_R011_T19TCJ_20220206T202105_resampled_subset.data
the data file is no longer generated. This is the most important for my application.
BEAM-DIMAP output consists of a .dim file with metadata, and a .data directory with each band stored as <name>.hdr (metdata) and <name>.img (image data). With GeoTIF you get one file with (often incomplete) metadata and all the bands (often with names like BAND1, etc.).
It may help to know more about the overall process. Are you using GNU parallel, a looping shell script, …?
So it is not an issue with the output format. You say it works for a “few runs”. I would make a note of the files that fail and try running them. If they fail reliably then something about their contents may be breaking gpt. If they run then we need to consider some sort of resource problem. In principle, each run of gpt is independent of other runs, but in practice there are optimizations that might cause the problem.
One optimization in linux is to delay removing programs from memory so they can be run in a loop with low overhead. Another is caching read and write data, both by the system and storage devices. CPU’s have been getting much faster but data storage has not kept pace. As a result, you can encounter situations where storage devices can’t keep up, which then requires more data to be cached by the system. Ideally the program generating data would be notified to slow down, but this is a rare cases so testing may not cover all situations. I would try adding a delay (in a shell script loop sleep can be used) between each gpt invocation.
For this sort of processing I have relied heavily on GNU parallel.
good morning,
I am running a serire of S2 images in a bash script calling the gpt in sequence. I am running in parallel but in sequence.
here is what I have this morning for a single job launched yesterday evening:
Not sure what this means. The GNU parallel program monitors system resources and can run shell scripts in parallel on a large system. Do you use parallel -j 1 to run jobs sequentially?
It is less than helpful to post images because they often omit key details such as the command line use to run a script. I can’t tell whether gpt is running in foreground or background. It takes more effort to quote sections from an image, and the contents are not searchable, so the next person who encounters the issue may not find this topic using a search.
Did the job produce the expected output?
When a shell script run in “foreground” finishes you should get back to the command prompt. If you run the script in background ( either by adding an ampersand (&) at the end of the command line or using and then the bg command) then you have the command prompt while the job is running. In workshops with users new to linux I have often had users reporting problems similar to yours who had multiple copies of a script in background because they confused <Ctrl-z> with <Ctrl-c>.
Depending on the linux distribution, there are many gui and command-line tools to view the status of running jobs along with memory usage. bpytop is widely available and gives a good view of the system status.
sorry, that was not clear indeed.
so, no I do not run my jobs in parallel.
yes the job created the expected output but the script did not go next job.
I know the difference between ctrl-z and ctrl-c and this is not the problem here.
how can I progress with that issue?
We need a lot more detail to understand your issue. It may help to post your shell script. Do you run it from the shell prompt in a terminal? Are you running scripts on a remote system or locally? Are the data files stored locally or using a network drive? DId the script return to the command prompt or was it stalled? Have you checked the output to make sure it is complete? Does your script check the return code ($?) for the gpt process?
When developing a gpt workflow it is useful to include the -e option to get more detailed error reporting.
You may want to surround the line with the gpt invocation with some progress reporting:
echo "gpt starting at $(date)"
gpt -e ...
echo "gpt finished with status $? at $(date)"
If you are running gpt on a remote system (e.g., using ssh from the local system to the remote) the session may be interrupted by network glitches. In such cases the nohup command is useful. It redirects the output of your job to a nohup.out file and will continue processing even if the terminal disconnects. It is good practice to monitor CPU loads, temperatures, memory usage, and disk usage while developing gpt workflows.
thanks for the tip.
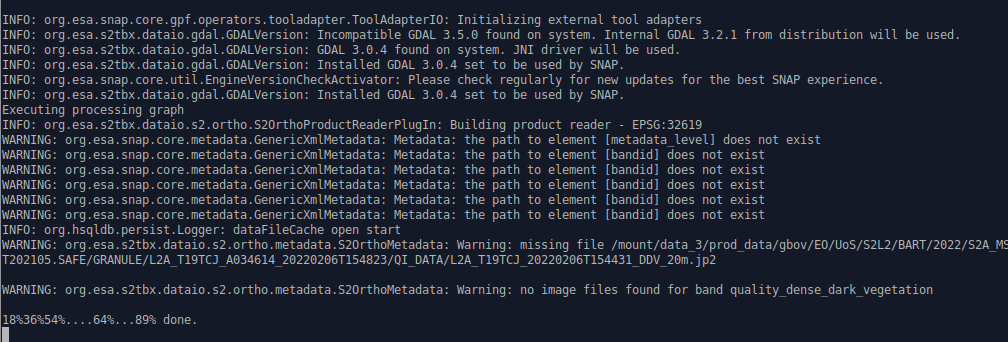
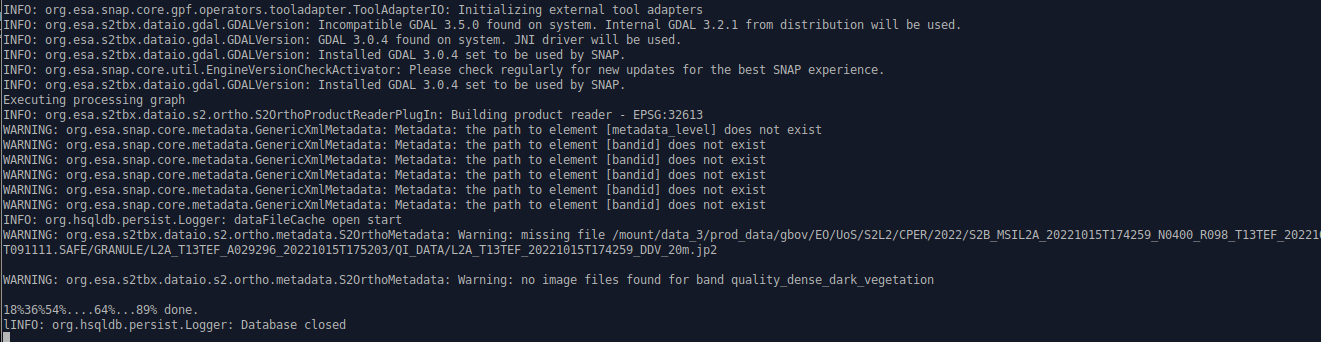
I lauched my script yesterday evening with the “-e” option.
It stuck but with no additional information:
INFO: org.esa.snap.core.gpf.operators.tooladapter.ToolAdapterIO: Initializing external tool adapters
INFO: org.esa.s2tbx.dataio.gdal.GDALVersion: Incompatible GDAL 3.5.0 found on system. Internal GDAL 3.2.1 from distribution will be used.
INFO: org.esa.s2tbx.dataio.gdal.GDALVersion: GDAL 3.0.4 found on system. JNI driver will be used.
INFO: org.esa.s2tbx.dataio.gdal.GDALVersion: Installed GDAL 3.0.4 set to be used by SNAP.
INFO: org.esa.snap.core.util.EngineVersionCheckActivator: Please check regularly for new updates for the best SNAP experience.
INFO: org.esa.s2tbx.dataio.gdal.GDALVersion: Installed GDAL 3.0.4 set to be used by SNAP.
Executing processing graph
INFO: org.esa.s2tbx.dataio.s2.ortho.S2OrthoProductReaderPlugIn: Building product reader - EPSG:32613
WARNING: org.esa.snap.core.metadata.GenericXmlMetadata: Metadata: the path to element [metadata_level] does not exist
WARNING: org.esa.snap.core.metadata.GenericXmlMetadata: Metadata: the path to element [bandid] does not exist
WARNING: org.esa.snap.core.metadata.GenericXmlMetadata: Metadata: the path to element [bandid] does not exist
WARNING: org.esa.snap.core.metadata.GenericXmlMetadata: Metadata: the path to element [bandid] does not exist
WARNING: org.esa.snap.core.metadata.GenericXmlMetadata: Metadata: the path to element [bandid] does not exist
WARNING: org.esa.snap.core.metadata.GenericXmlMetadata: Metadata: the path to element [bandid] does not exist
INFO: org.hsqldb.persist.Logger: dataFileCache open start
WARNING: org.esa.s2tbx.dataio.s2.ortho.metadata.S2OrthoMetadata: Warning: missing file /mount/data_3/prod_data/gbov/EO/UoS/S2L2/CPER/2022/S2B_MSIL2A_20221015T174259_N0400_R098_T13TEF_20221016T091111.SAFE/GRANULE/L2A_T13TEF_A029296_20221015T175203/QI_DATA/L2A_T13TEF_20221015T174259_DDV_20m.jp2
WARNING: org.esa.s2tbx.dataio.s2.ortho.metadata.S2OrthoMetadata: Warning: no image files found for band quality_dense_dark_vegetation
18%36%54%....64%...89% done.
INFO: org.hsqldb.persist.Logger: Database closed